In Course of implementation or validation of the computer system/ software with legal landscape of the Life Science industry, we have observed lot of misconception with respect to Computer System Validation. Let us understand the misconception and potential reasons:
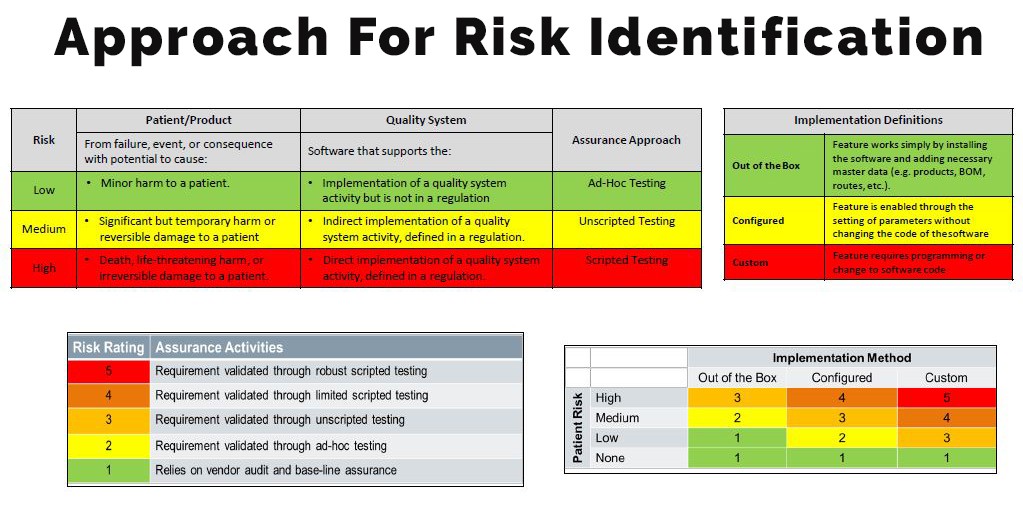
Identifying Risk potency
Most of the manufacturers misinterpret the criticality of application based on the direct or indirect impact (Risk) of software on the quality of product and patient safety.
Neoness #1 The current concept for software validation comes from 2002 and focuses on software which is embedded in device. However, now a days, there are many softwares available supporting the quality system of the manufacturer, however, they are not part of the device itself. The FDA has released a new draft guidance basis true risk-based approach, which should be considered when deploying indirect impacting non-product, manufacturing, operations, and quality system software solutions such as DMS, LMS, and QMS applications i.e. indirect impact on Quality.
Evidence Capture Method
The players in the ecosystem have been observed to emphasize producing various software documents like specification, test plans and test results along with evidence. The focus here lacks the Software assurance.
Neoness #2 Now a days most of the manufacturers are applying 21 CFR 820.70(i) irrespective of utility, operations or functions that directly or indirectly affect product safety or quality. However, FDA’s focus is to review validation of softwares that directly affects software assurance.
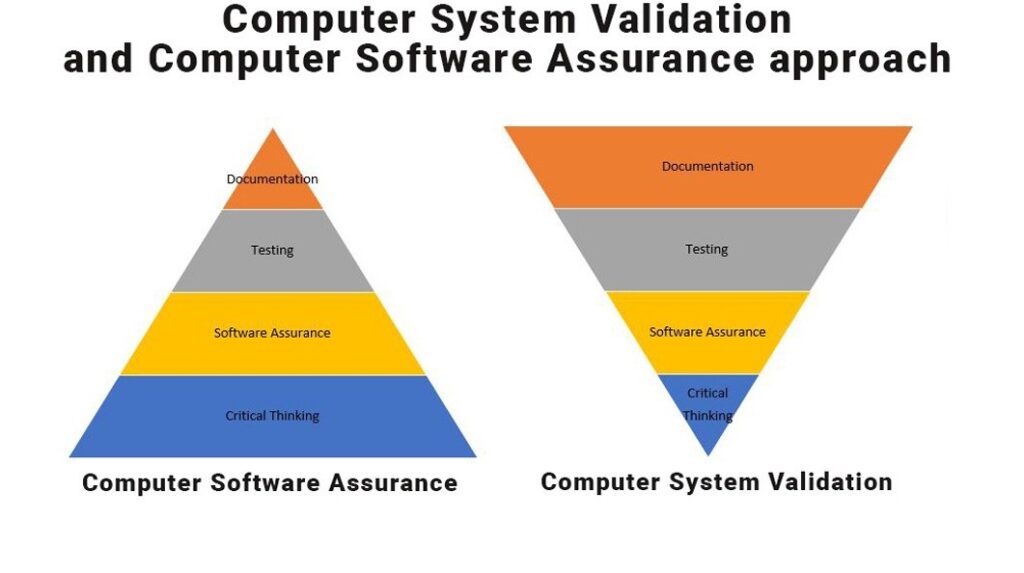
Test Method and Software Assurance:
Traditionally GAMP5 have become a simple checklist for Risk based approach to validate computer system in many companies (Trend captures last 10 years). Due to these guidelines, the manufacturers are more focusing on typical documentation i.e. as per “V” shape validation document requirement for all type of GxP softwares.
Neoness #3 New draft guidance will replace the term of “Computer System Validation” with “Computer Software Assurance”. This will guide the manufacturers to achieve true risk-based methodologies and will aid the manufacturer to achieve the software assurance to the best of it, during the SDLC process.
In order to provide a better clarity, FDA CDRH has released a draft guideline, making a shift from CSV to CSA for Manufacturing, Operations, and Quality System Softwares. In this draft guideline FDA has proposed the following to adopt elementary approach of software assurance:
Leverage of referring supplier data/ documentation in case software is indirectly impacting on product Quality.
21 CFR Part 11 narrowly scoped and is under enforcement discretion apply appropriately.
Go for automation Testing.
Using agile testing methods, unscripted exploratory and Ad hoc testing as appropriate.
Scripted testing (Limited and Robust testing) for higher software assurance.
Drug Manufacturers do not need to wait for this guidance to shift their mind-set towards Software Quality Assurance. They must be throttling up implementation of GXP softwares by adapting to CSA.